Dieser Praktikumsversuch ist der erste Teil eines dreiteiligen Blocks von Versuchen im Rahmen des Praktikums zur Molekülspektroskopie (S1: Quantenchemie auf dem Rechner; S2: Molekülspektroskopie auf dem Rechner; S3: Rotationsschwingungsspektren), in denen die Verzahnung der Theoretischen und der Physikalischen Chemie praktisch gezeigt werden soll.
Das Konzept besteht in Folgendem:
Im ersten Teil des Praktikums sollen Sie Punkte auf der Potentialkurve zweiatomiger Moleküle mit den Methoden der ab-initio-Quantenchemie ermitteln, um den Gleichgewichtsabstand herauszufinden.
Im zweiten Teil werden diese Punkte genutzt, um eine Morsefunktion zu erzeugen, die die Potentialkurve näherungsweise beschreibt. Basierend auf der Morsefunktion werden die Schwingungskonstante \(\omega_e\) und die Anharmonizitätskonstante \(\omega_ex_e\) ermittelt; daraus lässt sich zusammen mit der in Versuch S1 bestimmten Gleichgewichtsabstand das Rotations-Schwingungsspektrum prognostizieren.
Im dritten Teil sollen Sie das Rotationsschwingungsspektrum eines der berechneten Moleküle selbst aufnehmen und mit den Vorhersagen aus der Theorie vergleichen.
Quantenchemische Grundlagen
Die erfolgreiche Durchführung dieses Versuches setzt elementare Kenntnisse aus dem Bereich der molekularen Quantenmechanik voraus, die in der Vorlesung PC-II: »Physikalische Chemie II - Atombau und chemische Bindung« vermittelt werden.
Die Grundlagen der Quantenchemie können in dieser Skripte nur kurz genannt werden und sind in der Lehrbuchliteratur ausführlich beschrieben [1][2].
In der Quantenchemie werden die Methoden der Quantenmechanik zur
Beschreibung der Eigenschaften von Atomen und
Molekülen genutzt.
Quantenchemische Berechnungen stationärer Zustände molekularer Systeme werden durch
die Lösung der Schrödinger-Gleichung (SG),
\begin{equation}
\mathcal{H}(r,R) \Psi(r,R) = E \Psi(r,R)
\end{equation}
ermittelt, wobei \(\mathcal{H}(r,R)\) der Hamilton-Operator ist, der
das betrachtete, molekulare System beschreibt. \(r\) und
\(R\) sind jeweils die Koordinaten der Elektronen und der Kerne.
Die SG ist nur für wenige Systeme exakt lösbar, so dass verschiedene Näherungen notwending
sind.
Eine wichtige Vereinfachung erfolgt durch die Born-Oppenheimer-Näherung
(BO). Aufgrund des sehr großen
Masseunterschiedes zwischen Atomkernen und Elektronen
bewegen sich Elektronen sehr viel schneller als Kerne, und
es ist sinnvoll anzunehmen, dass sich die Elektronen unverzüglich jeder beliebigen Anordnung der Kerne anpassen.
Kern- und Elektronenbewegung sind also separierbar.
Für die SG bedeutet dies, dass sich der Hamilton-Operator,
\(\mathcal{H}(r,R)\), in eine Summe aus einem Operator für
die Kerne, \(\mathcal{H}_K(R)\), und einem für die Elektronen
und deren Wechselwirkung mit den Kernen, \(\mathcal{H}_e(r,R)\),
additiv zerlegen lässt:
\begin{equation}
\mathcal{H}(r,R) = \mathcal{H}_K(R) +\mathcal{H}_e(r,R).
\end{equation}
Die beiden Beiträge lassen sich darstellen als
\begin{equation}
\mathcal{H}_K(R) = \mathcal{T}_K(R) + \mathcal{V}_{KK}(R)
\end{equation}
und
\begin{equation}
\mathcal{H}_e(r,R) = \mathcal{T}_e(r) + \mathcal{V}_{ee}(r,R)
+ \mathcal{V}_{Ke}(r,R).
\end{equation}
\(\mathcal{T}_K(R)\) und \(\mathcal{T}_e(r)\) sind jeweils die
kinetischen Energien der Kerne und der Elektronen.
\(\mathcal{V}_{KK}(R)\), \(\mathcal{V}_{ee}(r,R)\) und
\(\mathcal{V}_{Ke}(r,R)\) beschreiben die Wechselwirkung
zwischen Elektronen, zwischen Kernen sowie zwischen Kernen
und Elektronen.
Die Wellenfunktion \(\Psi(r,R)\) läßt sich dementsprechend als
Produkt aus zwei Funktionen \(\Psi_K(R)\) und \(\Psi_e(r,R)\) beschreiben,
wobei \(\Psi_K(R)\) ausschließlich von der Position \(R\) der Kerne,
aber \(\Psi_e(r,R)\) (sowie \(\mathcal{V}_{ee}(r;R)\) und \(\mathcal{V}_{Ke}(r,R))\))
von der Position der Kerne und der Elektronen, \(R\) und \(r\), abhängt.
Für den Fall ruhender Kerne (BO Näherung!!) ist \(\mathcal{T}_K(R)=0\)
und \(\mathcal{V}_{KK}(R)=\text{const.}\) (elektrische Abstossung der Kerne)
und man erhält für die SG
\begin{equation}
(\mathcal{H}_e(r,R) + \mathcal{V}_{KK}(R)) \Psi_e(r,R) \Psi_K(R)
= E(R) \Psi_e(r,R) \Psi_K(R).
\label{sep-sch}
\end{equation}
Da \(\mathcal{V}_{eK}(r,R)\) ein multiplikativer Operator ist, läßt
sich (\ref{sep-sch}) als
\begin{equation}
\Psi_K(R) \mathcal{H}_e(r,R) \Psi_e(r,R) + \mathcal{V}_{KK}(R)
\Psi_e(r,R) \Psi_K(R) = E(R) \Psi_e(r,R) \Psi_K(R)
\end{equation}
schreiben, und man erhält schließlich
\begin{equation} \label {ElecSG}
\mathcal{H}_e(r,R) \Psi_e(r,R) = (E(R) - \mathcal{V}_{KK}(R)) \Psi_e(r,R).
\end{equation}
Gl. \ref{ElecSG} wird als elektronische SG bezeichnet und ihre
Eigenwerte sind nur noch parametrisch von den Kernpositionen
abhängig: für einen gegebenen Satz von festgehaltenen Kernkoordinaten lässt sich die Energie des Moleküls ausrechnen. Für einen anderen Satz von Koordinaten erhält man eine andere Energie. Die Kernkoordinaten treten nicht mehr als Variable auf.
Die Gesamtheit der Punkte, die sich bei Variation der Kern-Koordinaten durch jeweilige Lösung von Gl. \ref{ElecSG} ergibt, wird als die Potentialenergiefläche eines Moleküls bezeichnet. Sie spielt eine
wichtige Rolle zum Beispiel bei der Untersuchung chemischer Reaktionen
und der Molekülschwingungen in der IR-Spektroskopie.
Für zweiatomige Moleküle lassen sich die Kernkoordinaten durch den Abstand der Kerne wiedergeben, da die absolute Lage des Moleküls im Raum nicht interessiert (wenn er feldfrei ist). Aus der Potentialenergiefläche wird damit die Potentialkurve. Die Potentialkurve
wird demnach punktweise ermittelt, wobei jeder Punkt der Lösung
der elektronischen SG (Gl. \ref{ElecSG}) für einen Abstand der Kerne \(R\) entspricht.
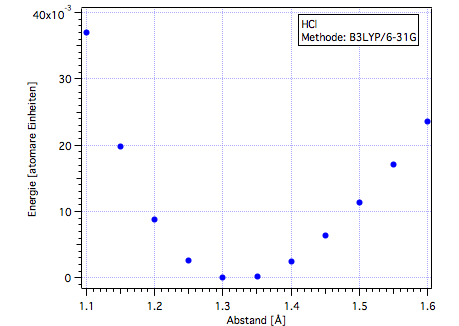
Ein Beispiel für quantenchemisch berechnete Wertepaare Abstand\(\to\)Energie ist in der Abbildung 1 gezeigt. Beachten Sie bitte, dass die Punkte unverbunden sind, da nur die Einzelpunkte berechnet sind, aber keine (stetige) Kurve aus quantenmechanischen Rechnungen im Rahmen der BO-Näherung ermittelt werden kann.
Abb. 1: Beispiel einer quantenchemischen Berechnung der Energie des Chlorwasserstoff-Moleküls (HCl) für unterschiedliche Kernkoordinaten in der Nähe des Gleichgewichtsabstandes. Die Energie enthält nicht die kinetische Energie der Kerne, da diese im Rahmen der Born-Oppenheimer-Näherung als ruhend angenommen werden. Für die Energie wird die atomare Einheit (Hartree) verwendet: 1 Hartree = 27,2114 eV. Das Hartree ist eine Lieblingseinheit der Theoretiker, in der Physikalischen Chemie und Spektroskopie werden eher das Elektronenvolt (eV) und die Wellenzahl als Energie-proportionale Einheit verwendet.
Orca
Alle Rechnungen in diesem Versuch werden mit dem Programmpaket
Orca durchgeführt. Orca ist ein großes
Programmsystem zur Berechnung der elektronischen Struktur von
Molekülen mit Hilfe von ab-initio-Methoden (Hartree-Fock, CI, CC,
MPn, etc.), einigen semi-empirischen Verfahren sowie der
Dichtefunktionaltheorie (DFT). Darüber hinaus gestattet es die
Berechnung von Schwingungsfrequenzen, thermodynamischen Eigenschaften,
Abschirmkonstanten für die NMR-Spektroskopie, Übergangszuständen
und Lösungsmitteleffekten. Da es (relativ) übersichtlich und
einfach zu bedienen ist, ermöglicht es vor allem auch
Nicht-Theoretikern einen Zugang zu quantenchemischen Berechnungen und
damit zur quantitativen Beschreibung
molekularer Strukturen und chemischer Reaktionen.
Außerdem ist das Open Source Programm für die nicht-kommerzielle Verwendung für alle frei zugänglich.
Um eine quantenchemische Rechnung mit Orca
durchzuführen, ist es zunächst nötig, eine sog. Input-Datei zu
erzeugen.
Beispiel-Inputs für das Orca-Programm finden Sie in der
Orca Input Library.
Inputs haben einen programmspezifischen Syntax:
# Das ist ein Kommentar
! rohf def2-tzvp
*xyz 0 1
C 0.0 0.0 0.0
O 0.0 0.0 1.1030
*
Nach einem ! folgen einfache Keywords.
Am Keyword ROHF erkennt das Programm, dass Hartree Fock verwendet werden soll.
Nach einem * erfolgt die Eingabe der geometrischen Struktur als Element + Kartesische Koordinaten (xyz).
0 1 beziehen sich auf eine neutrale Ladung und Singlet-Multiplizität (Entartung, 2S+1).
Die Moleküle können mit Programmen wie
molden, avogadro oder chemcraft visualisiert werden.
Dabei kann auch die elektronische Struktur (wie z. B. Molekülorbitale) visualisiert werden.
Qualität der Rechnungen
Die erreichte Genauigkeit von quantenchemischen Rechnungen wird im
wesentlichen von zwei Faktoren bestimmt:
Verwendete Methode:
Es gibt eine Vielzahl verschiedener quantenchemischer Methoden, die
mit verschiedenen Ansätzen und Aufwand versuchen, die Lösung des
elektronischen Problems zu nähern.
Dabei unterscheidet man zwischen
Methoden, die auf Näherungen für die elektronische Wellenfunktion
basieren wie Hartree-Fock (HF), darauf aufbauende
Methoden, und der Dichtefunktionaltheorie (DFT).
Verwendeter Basissatz:
Quantenmechanische Systeme werden mit Hilfe von Basisfunktionen beschrieben.
Basisfunktionen sind Atomorbitale.
Prinzipiell gibt Basisfunktionen für s-Orbitale, p-Orbitale, d, f, g, h ...
Für eine exakte Beschreibung sind unendlich viele Basisfunktionen notwendig.
Je mehr Basisfunktionen man benutzt, desto besser ist das Ergebnis,
gleichzeitig steigt aber auch der Rechenaufwand an.
Für eine Lösung des Problems muss eine endlich Menge davon ausgewählt werden.
Vorbereitung
Bringen Sie einen USB-Stick oder Cloud-Zugang mit um die Daten abzuspeichern.
Fassen Sie kurz zusammen, wie man quantenchemisch die Potentialfläche bzw. -kurve für ein Molekül erhält.
Zeichnen Sie als Vorbereitung ein MO-Diagramme für CO.
Suchen Sie nach Daten für Dissoziationsenergie und Bindungsabstand von CO (Temperaturabhängigkeit beachten).
Wir werden auf einem Linuxcomputer arbeiten.
Nehmen Sie sich 20 Minuten Zeit um die folgenden Einführungsvideos für die Kommandozeile auf Linux anzusehen.
Linux Shell and Command Line,
Files and Directories
Es braucht viel Übung um gut mit der Kommandozeile umgehen zu lernen, weshalb wir uns näher damit auseinandersetzen werden sobald ein Terminal zur Verfügung steht.
Aufgaben während des Praktikumsversuches
(Bitte achten Sie darauf, für jede Rechnung eine neue Inputdatei zu erstellen.
Sie können die alten Dateien mit cp old.inp new.inp dublizieren.)
Beginnen Sie mit einer Rechnung für \(\ce{CO}\).
Öffnen Sie dazu mit gedit co_hf.inp eine Datei und schreiben Sie den oben beschriebenen Input ab.
Verwenden Sie den Befehl orca co_hf.inp > co_hf.out um die Rechnung zu starten.
Machen Sie sich mit der Ausgabedatei vertraut.
Ist die Optimierung der elektronischen Struktur konvergiert?
Wie viele Iterationen hat das Self Consistent Field SCF benötigt um die elektronische Struktur zu optimieren?
Suchen und notieren Sie sich die FINAL SINGLE POINT ENERGY!
Berechnen Sie nun die Dissoziationsenergie des Moleküls.
ΔE = EC + EO - ECO
Sie benötigen dazu die Energien der Atome C und O im jeweiligen elektronischen Grundzustand \(^3{\rm P}\).
Da wir eine homogene Dissoziation berechnen wollen bleibt die Ladung \(q\) der isolierten Atome: \(q= 0\).
Erstellen Sie die Input Dateien der Atome.
Vergleichen Sie die Dissoziationsenergie mit experimentellen Daten aus der Literatur.
Neben der Gesamtenergie des Moleküls liefern die quantenchemischen Rechnungen auch die Molekülorbitale (Eigenvektoren) und deren Energien (Eigenwerte).
Suchen Sie nach ORBITAL in den Outputs.
Zeichnen Sie fürs Protokoll ein detailliertes MO-Diagramm für besetzte Orbitale.
Wie Sie an Ihrem Ergebnis feststellen werden, ist Hartree Fock keine geeignete Methode, um die Dissoziationsenergie zu berechnen.
Versuchen Sie es erneut mit dem Dichtefunktional PBE0.
Erkunden Sie die Potentialkurve und
finden Sie die optimale Bindungslänge durch Variation des Abstandes heraus.
Variieren Sie die Bindungslänge und berechnen Sie zusammen insgesamt 30 Punkte.
Erstellen Sie eine Wertetabelle mit den Abstand-Energie Wertepaaren.
Die Wertepaare werden für den Versuch S2 benötigt.
Verwenden Sie nun das Keyword "OPT" um die geometrische Struktur zu optimieren.
Wieviele Iterationen braucht die geometrische Strukturoptimierung?
Ist die geometrische Strukturoptimierung konvergiert? (Keyword: HURRAY)
Welchen Wert hat der Gleichgewichtsabstand?
Vergleichen Sie die von Ihnen ermittelten Bindungsabstände mit experimentellen Daten aus der Literatur.
Fürs Protokoll: Stellen Sie Ihre Ergebnisse in geeigneten Graphiken mit korrekter
und vollständiger Achsenbeschriftung sowie in tabellarischer Form dar.
Achten Sie darauf, die Energie so zu verschieben,
dass das Minimum der »Kurve« gleich Null ist.
Der niedrigste Energiewert muss von allen anderen abgezogen werden.
Konvertieren Sie die Einheit von Hartree nach kJ/mol.
Zusätzliche Aufgaben
Berechnen Sie die Dissoziationsenergie für \(\ce{CO}\) mit einem endlichen Abstand (z.B. 10000 Å).
Man könnte nun annehmen, dass die Wechselwirkung zwischen den Atomen null ist und das Resultat der Dissoziationsenergie gleich sein muss.
Warum sind bei endlichen Abständen HF und DFT keine geeigneten Methoden um die Dissoziation des \(\ce{CO}\)-Moleküls zu beschreiben?
Welche Methoden könnte man verwenden um die Dissoziation der Atome auch bei endlichen Abständen korrekt zu beschreiben?
Literatur
[1] W. Kutzelnigg, Einführung in die Theoretische Chemie, Wiley-VCH, Weinheim 2001.
[2] T. Engel, P. Reid, Physikalische Chemie, Kapitel 27, San Francisco: Pearson Benjamin Cummings 2006.