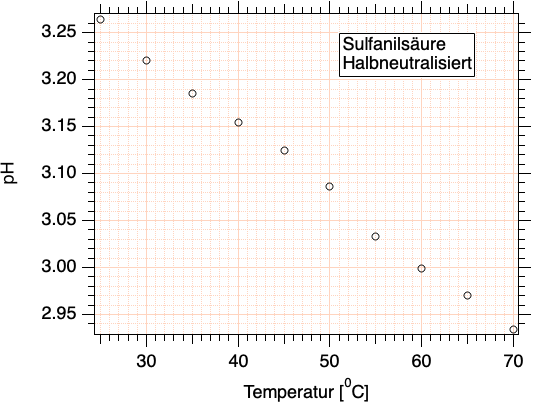
Bestimmung des pH-Wertes von halbdissozierter Sulfanilsäure bei Raumtemperatur sowie bei Temperaturen zwischen 25 \(^0\)C und 70 \(^0\)C.
Ermittlung des \({\rm pK_{S}}\)-Wertes der Sulfanilsäure als Funktion der Temperatur unter Berücksichtigung der Ionenstärke.
Berechnung der Dissoziationsenthalpie aus der
Temperaturabhängigkeit des pK-Wertes mittels der van’t-Hoffschen Gleichung.
Stichworte zur Vorbereitung
Temperaturabhängigkeit der Gleichgewichtskonstanten
Dissoziationsgleichgewichte
Mittlere Ionenaktivität und mittlerer Aktivitätskoeffizient
Aktivitätskoeffizient und Ionenstärke
pH-Wert und pH-Wert-Messung
Theoretische Grundlagen
1. Die van't Hoffsche Gleichung
Für eine chemische Gleichgewichtsreaktion mit der Gleichgewichtskonstanten \(K\) und der freien Standard-Gibbs-Enthalpie \(\Delta G^0\) gilt:
\[
K
= \exp\left(- \frac{\Delta G^0}{RT}\right) = \exp \left(- \frac{\Delta H^0 - T \cdot \Delta S^0}{RT} \right)
= \exp \left( \frac{- \Delta H}{R}\frac{1}{T}\right) \cdot \exp \left( \frac{\Delta S}{R} \right)
\]
Logarithmieren liefert:
\[
\ln K = \frac{- \Delta H}{R}\cdot \frac{1}{T} + \frac{\Delta S}{R}
\]
Fassen wir die reziproke Temperatur \(\dfrac{1}{T}\) als unabhängige Variable (\(1/T\) ist sozusagen x, und \(\ln K\) ist y) auf, dann gilt
Bei der undissoziierten Säure HA kann die Konzentration gleich der Aktivität gesetzt werden. Beim Anion \(\ce{A-}\) ist das nicht der Fall.
Die Aktivität wird als Produkt aus der Konzentration \(c\) und dem Aktivitätskoeffizienten \(f_{\rm}\) dargestellt (zu \(f_{\rm}\) siehe weiter unten):
\[
a(\ce{A-}) = c(\ce{A-}) \cdot f_{\pm}
\]
Daraus folgt
\[
pK_{S} = pH - \log \frac{c(\ce{A-}) \cdot f_{\pm}}{c(\ce{HA})}
\]
Neutralisiert man 50% der Säure mit einer starken Basen, dann gilt
\[
\frac{c(\ce{A-}) }{c(\ce{HA})} = 1,
\]
so dass verbleibt:
\begin{equation} \label {eqpKS1}
pK_S = pH - \log f_{\pm}.
\end{equation}
Für \(\log f_{\pm}\) nehmen wir an (wir stellen diesen Wert experimentell ein, siehe weiter unten):
\[
\log f_{\pm} = -0.122
\]
\[
pK_S = pH + 0.122.
\]
Berücksichtigt man die Aktivitäten, dann gilt bei Halbneutralisation also nicht länger die bekannte Gleichung \(pH=pK_S\).
Zahlenbeispiel für \(25\;^0C\):
Es wird ein pH=3.264 gemessen. Dann ist mit \(\log f_{\pm} = -0.122\):
\[
pK_S = pH + 0.122 = 3.264 + 0.122 = {\bf 3.386}.
\]
3. Einfluss der Temperatur auf das Säure-Base-Gleichgewicht
Wenn die Reaktionsenthalpie des Prozesses in Gl. \ref{eqDissoz} von Null verschieden ist, dann ändert sich gemäß Gl. \ref{eqvantHoff} die Gleichgewichtskonstante \(K\) und somit auch der \(pK_S\)-Wert der Säure.
Bei einer Temperaturerhöhung reagiert das System, indem entweder ein Anteil der nicht-dissoziierten Säure HA nachdissoziiert (folglich gibt es dann mehr \(\ce{A-}\) und gerade soviel weniger \(\ce{HA}\), wie es mehr \(\ce{A-}\), oder umgekehrt ein Anteil von \(\ce{A-}\) wieder in die Form HA übergeht.
Den Anteil von \(a (A^-)\), um den sich die Aktivität des Anions ändert, nennen wir \(\Delta a (A^-)\).
Die Aktivität des Anions bei Temperaturerhöhung ändert sich also von \(a_0 (A^-)\) (Ausgangswert bei Halbneutralisation) auf \(a_0(A^-) + \Delta a (A^-)\). Die Größe \(\Delta a (A^-)\) kann dabei sowohl positiv (Nachdissoziation) als auch negativ (Rückbildung von HA) sein.
Demnach gilt bei einer höheren Temperatur (die nachfolgende Gleichung ist von größter Bedeutung):
\[
pK_S = pH - \log \frac{a_0(A^-) + \Delta a (A^-)}{c_0(HA) - \Delta a (A^-)}
\]
Da bei der Bildung von \(\Delta a (A^-)\) gerade so viele \(\ce{H3O+}\)-Ionen gebildet werden, wie sich \(\ce{A-}\)-Ionen aus HA bilden, können wir setzen:
\[
\Delta a (A^-) = \Delta a(\ce{H3O+})
\]
und damit:
\[
pK_S = pH - \log \frac{c_0(A^-)\cdot f_{\pm} + \Delta a (\ce{H3O+})}{c_0(HA) - \Delta a (\ce{H3O+})}
\]
Mit \(10^{-pH}=a(\ce{H3O+})\) folgt unmittelbar:
Rechenbeispiel: Sei \(T=50\;^0C\) und der pH-Wert wird experimentell zu pH = 3.086 bestimmt; ferner sei bei dieser Temperatur \(f_{\pm}= 0.744 \).
Mit \(c_0(A^-) = c_0(HA) = 0.005\) mol/L (experimentell eingestellt Werte, siehe unten) erhält man
\[
pK_{S_{50\,^0C}} = pH_{50\,^0C} - \log \frac{\left(0.005 \cdot 0.744\right) + \left(10^{-3.086} - 10^{-3.264}\right)} {0.005 - \left(10^{-3.086} - 10^{-3.264}\right)}
\]
Mit \(\left(10^{-3.086} - 10^{-3.264}\right) = 2.758 \cdot 10^{-4} \)
ergibt sich:
\[
pK_{S_{50\,^0C}} = 3.086 - \log \frac{\left(0.005 \cdot 0.744\right) + 2.758 \cdot 10^{-4}} {0.005 - 2.758 \cdot 10^{-4}} = 3.086 - (-0.0727245) = {\bf 3.159}.
\]
Der \(pK_S\)-Wert der Säure erniedrigt sich also bei Temperaturerhöhung von \(25\,^0{\rm C}\) auf \(50\,^0{\rm C}\) von \(pK_S=3.386\) auf \(pK_S=3.195\). Das sieht nicht nach viel aus, aber die Gleichgewichtskonstante \(K= 10^{-pK_S}\) ändert sich dabei immerhin um ca. 25%!
Bestimmung von \(\Delta H\)
Wir gehen aus von Gl. \ref{eqvantHoff}:
\[
\frac{\diff \ln K}{\diff 1/T} = \frac{-\Delta H}{R}.
\]
Nach Definition ist
\[
pK_S = - \log K_S
\]
Zum \(\ln K_S\) gelangen wir durch Multiplikation der linken Seite mit \(\ln(10\):
\[
pK_S \cdot \ln(10) = - \ln K_S,
\]
wobei
\[
\ln(10) = 2.303.
\]
Also gilt:
\[
pK_S \cdot 2.303 = - \ln K_S.
\]
Dann gilt auch:
\[
2.303 \cdot \diff \;pK_S = - \diff \ln(K_S)
\]
und durch Teilen durch \(\diff 1/T\):
\[
\frac{2.303 \cdot \diff \;pK_S}{\diff 1/T} = \frac{- \diff \ln(K_S)}{\diff 1/T} = \frac{\Delta H}{R}
\]
Bekanntlich (vgl. Lehrbücher der Elektrochemie) werden bei chemischen Gleichgewichtsreaktionen, bei denen geladene Teilchen auftreten, nicht die Konzentrationen dieser Teilchen genutzt, sondern vielmehr ihre Aktivität.
Diese lässt sich darstellen als das Produkt aus der Konzentration und einem Aktivitätskoeffizienten \(f\).
Es können aber nicht individuelle Aktivitätskoeffizienten, sondern lediglich mittlere Aktivitätskoeffizienten, die mit dem Symbol \(f_{\pm}\) bezeichnet werden.
Die mittleren Aktivitätskoeffizienten lassen sich mit der Debye-Hückel-Gleichung
\begin{equation} \label {Debye}
\log_{10}(f_{\pm}) = - A \cdot z_+ \cdot |z_-| \frac {\sqrt{I}}{\sqrt{1\; \rm mol/L}+\sqrt{I}}
\end{equation}
berechnen, wenn die Ionenstärke \(I\) bekannt ist. (\(I\): Ionenstärke in mol·\({\rm L}^{-1}\).)
Beachten Sie bitte, dass der Ausdruck \(\sqrt{1\;{\rm mol/L}}\) im Nenner der
Gl. \ref {Debye} die Zahl \(1\) enthält.
Die in der Gl. \ref{Debye} auftretende Ionenstärke \(I\) ist wie folgt definiert:
\begin{equation}
I = \frac {1}{2} \cdot \sum c_i z_i^2.
\end{equation}
\(c_i\) ist die Konzentration der i-ten Ionensorte, und \(z_i\) ist die Ladung eines Ions dieser Sorte.
Bei 1:1-Elektrolyten (Beispiel: NaCl) ist die Ionenstärke gleich der Salzkonzentration.
Der Index \(i\) bezieht sich auf alle Ionen in der Lösung, \(I\) ändert sich
also je nach dem Ausmaß der elektrolytischen Dissoziation mit der Temperatur; damit ändert sich dann aber auch \(f_{\pm}\), was die Datenanalyse problematisch macht.
Um die Ionenstärke konstant zu halten, wird Natriumchlorid in großem
Überschuss zur Lösung hinzu gegeben.
Im Temperaturintervall 25 \(^0\)C -70 \(^0\)C lässt sich die Konstante \(A\) aus der Gl. \ref{Debye} für
beliebige Temperaturen mittels der folgenden Gleichung bestimmen:
\begin{equation}
A = 0,48816 + 7,36 \cdot 10^{-4}\; T + 4,091 \cdot 10^{-6}\; T^2.
\end{equation}
In dieser Gleichung ist \(T\) die Temperatur in \(^0\)C (nicht in K).
Praktische Durchführung der Messungen
Die Apparatur besteht aus einem doppelwandigen temperierbaren Gefäß, das an einen Thermostaten angeschlossen ist, einer pH-Messelektrode (Sensor) sowie einem Auslesegerät für den pH-Sensor. Der Elektrolyt wird während der Messungen mit einem Magnetrührer gerührt.
Die empfindliche pH-Messelektrode wird vor den Messungen in einem Gefäß mit gesättigter KCl-Lösung aufbewahrt, um sie vor Schaden zu bewahren. Sie ist vor dem Einsetzen in die Meßzelle gründlich mit dreifach destilliertem Wasser zu spülen und nach Beendigung der Messung wiederum zu spülen und nachfolgend in das Aufbewahrungsgefäß zu hängen.
Verwenden Sie zur Lagerung kein destilliertes (= entionisiertes) Wasser aus der Laborversorgung, da dieses die Elektrode schädigt.
Versuchsdurchführung im Einzelnen
1. Stellen Sie sich durch Mischen von je 50 ml 0.01-M NaOH und 0.02-M Sulfanilsäure eine Lösung her, die äquimolare Mengen an freier Sulfanilsäure und ihrem Natriumsalz enthält (warum ist jetzt die Konzentration des Sulfanilsäure-Anions gleich 0.005 mol/L?). Verwenden Sie hierzu die zur Verfügung gestellten Pipetten.
2. Stellen Sie durch Einwägen von festem NaCl in der Lösung eine Ionenstärke I von ca. 0.1-M ein. Die Berechnung steht weiter unten.
Es kommt nicht darauf an, dass die Ionenstärke ganz genau gleich 0,1-M ist, sondern vielmehr darauf, dass Sie die Masse des NaCl genau kennen und daraus die genaue Ionenstärke berechnen können. Dabei ist neben den Beiträgen aus dem NaCl auch der Beitrag des Natriumsalzes der Sulfanilsäure zu berücksichtigen (Kation und Anion!).
3. Lesen Sie diesen Punkt zunächst ganz durch! Messen Sie im Temperaturintervall 25-70 \(^0\)C den pH-Wert der Lösung. Die bereitgestellten Thermostaten verfügen über einen internen Kühlkreislauf, sie brauchen nicht ans Leitungswasser angeschlossen zu werden. Die Solltemperatur kann am Thermostaten digital eingestellt werden.
Messen Sie den pH-Wert der Messlösung in 5\(\;^0\)C-Schritten. Die relevante Temperaturmessung erfolgt in der Messlösung, nicht im Thermostaten! Warten Sie nach dem Erreichen einer konstanten Temperatur in der Messlösung noch ca. 1 Minute, bevor Sie den pH-Wert ablesen, um thermisches Gleichgewicht zu gewährleisten! Tragen Sie die Tempertur und den zugehörigen pH-Wert in eine Tabelle ein.
Zur Vermeidung von Missverständnissen: es ist nicht wichtig, dass Sie die Temperatur jeweils um exakt 5 Grad erhöhen. Es kommt nur darauf an, dass Sie die Temperatur, bei der aktuell gemessen wird, genau ablesen.
Die auftretenden Änderungen des pH-Wertes sind klein - der pH ändert sich um weniger als 0,5 Einheiten. Ausreichend genaue Ergebnisse erhalten Sie daher nur, wenn Sie sorgfältig arbeiten.
Auswertung
Ermitteln Sie hieraus den jeweiligen pK\(_{\rm S}\)-Wert bei den einzelnen Messtemperaturen durch Nutzung der Gl. \ref{eqMessgleichung}
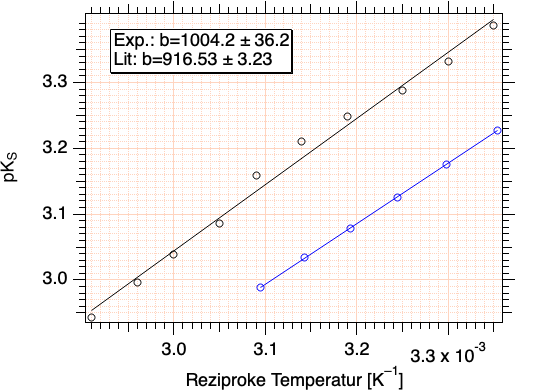
Tragen Sie die ermittelten pK\(_{\rm S}\)-Werte gegen die reziproke absolute Temperatur in K\(^{-1}\) auf und ermitteln Sie die Ausgleichsgerade.
Bestimmen Sie durch Nutzung der Gl. \ref{eqGetDeltaH} aus dem Anstieg der gefundenen Ausgleichsgeraden die Standard-Reaktionsenthalpie \(\Delta\)H\(^0\) für die elektrolytische Dissoziation der Sulfanilsäure bei der genutzten Ionenstärke.
Zeigen Sie, auf welche Weise Sie aus Ihrer Einwaage an KCl die Ionenstärke in der Messlösung berechnet haben!
Zur Bestimmung der Ionenstärke
Wir bestimmen im Folgenden die Menge an NaCl, die hinzugefügt werden muss, um insgesamt eine Ionenstärke von 0.1 mol/L einzustellen.
Nach Hinzufügen der NaCl zur halbdissoziierten Sulfanilsäure befinden sich folgende Ionen in der Lösung:
Na\(^+\) aus NaCl;
Cl\(^-\) aus NaCl;
Na\(^+\) aus NaOH;
Sulfanilatanionen (Sulf\(^{-}\)) aus der Sulfanilsäure;
H\(^+\)-Ionen;
OH\(^-\)-Ionen.
Für die Ionenstärke gilt:
\[
I = \frac {1}{2} \sum c_iz_i^2.
\]
Hier also (summiert über alle Ionen):
\[
I = \frac {1}{2} \left(c({\rm Na}^+)_{\rm Sulf} + c({\rm Sulf}^-) + c({\rm Cl}^-) + c({\rm Na}^+)_{\rm NaCl} + c({\rm H}^+) + c({\rm OH}^-)\right) \stackrel{!}{=} 0.1\;{\rm mol/L}
\]
Mit \(c({\rm Na}^+)_{\rm Sulf}\) ist die Konzentration an Na\(^+\) aus dem Natriumsulfanilat gemeint, das durch die Halbneutrealisation der Sulfanilsäure mit NaOH-Lösung gebildet wurde. Entsprechend bedeutet \(c({\rm Na}^+)_{\rm NaCl}\) die noch unbekannte Konzentration an Na\(^+\) durch Zugabe von NaCl.
Der pH-Wert beträgt zu Beginn der Messung \(\approx\) 3, entsprechend einer \(\ce{H+} \)-Ionen-Konzentration von 10\(^{-3}\) mol/L. Die OH\(^-\)-Konz. beträgt demnach etwa 10\(^{-11}\) mol/L. Die Konzentrationen an OH\(^-\) können wir daher im Folgenden vernachlässigen.
Da \(c({\rm Na}^+)_{\rm NaCl} = c({\rm Cl}^-)\) für NaCl und analog für das Natriusulfanilat, schreiben wir:
D
Die Konzentration der Sulfanilsäure beträgt zunächst 0,02 mol/L. Nach der Zugabe der 0,01-M NaOH liegt die Säure zu 50% dissoziiert vor, aber im doppelten Flüssigkeitsvolumen (jetzt zusammen 100 mL statt 50 mL), so dass die Konzentration an Sulfanilat-Anion nunmehr 0,005 mol/L beträgt. Da \( \frac{1}{2} c(\ce{H3O+}) \approx 0.0005 \) mol/L, wird dieser Term vernachlässigt. Daher:
\[c({\rm NaCl}) = (0,1 - 0,005) \; \frac {\rm mol}{\rm L} = 0,095 \; \frac {\rm mol}{\rm L}. \]
Wir benötigen 0.095 Mol/L NaCl. Da wir nur 100 mL Lösung vorliegen haben, benötigen wir nur 0.0095 mol NaCl.
1 mol NaCl = 58,44 g;
0.0095 mol NaCl \(\approx\) 0,555 g.
Wir benötigen 0,56 g NaCl.
Wenn wir Kaliumchlorid statt Natriumchlorid verwenden, müssen wir
berücksichtigen, dass die Molmasse von Kaliumchlorid zu M(KCl)=74,55 g/mol
gegeben ist. Es ergeben sich dann 0,71 g Kaliumchlorid.
Literatur
R. O. McLaren, D. F. Swinehart, J. Am. Chem. Soc. 73, 1822 (1951).
Perrin, D. D., Dissociation Constants of Organic Bases in Aqueous Solution, Butterworths, London, 1965; Supplement, 1972. Dieser Quelle entnehmen wir folgende
Literaturwerte bzgl. des pK\(_S\)-Wertes der Sulfanilsäure:
Temperatur / °C
\(pK_S\)
25
3,2273
30
3,176
35
3,126
40
3,0788
45
3,0339
50
2,9892
Ergebnisse (Beispiel)
Typische Ergebnisse eines Praktikumsversuches sind in den Abb. T4-1 (Rohdaten) und T4-2 (Auswertung) dargestellt.
Abb. T4-1: pH-Wert einer halbneutralisierten Sulfanilsäure bei unterschiedlichen Temperaturen (exp. Ergebnisse).
Abb. T4-2: Auswertung experimenteller Ergebnisse (schwarz) und Vergleich mit Literaturdaten (blau).
Version (Latex): 23.04.2019, Flesch
Version (html/MathJax): 12.03.2021, Flesch, Demirkaya
07.11.2024: erhebliche Änderungen, u.a. Ermittlung von \(\Delta S\) entfernt. (Flesch, Al Najjar)