Aufnahme der Absorptionsspektren dreier Cyanin-Farbstoffe
Experimentelle Ermittlung der Wellenlänge des Absorptionsmaximums
Bestimmung der Kettenlänge \(L\) der Cyanine
Beachten Sie bitte die zusätzlichen Aufgaben am Ende dieser Skripte!
Grundlagen
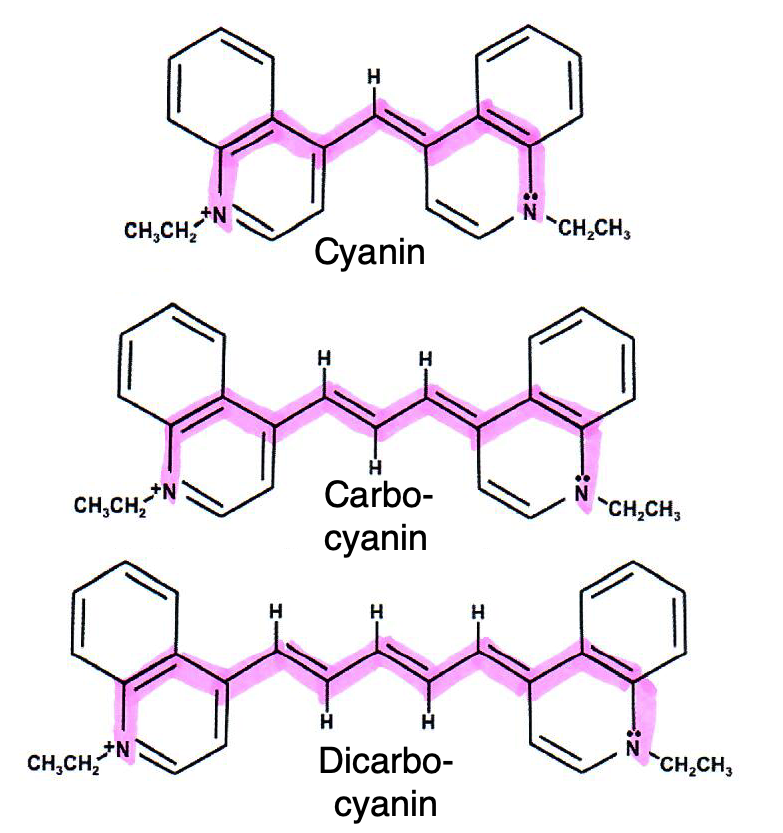
1.– Betrachten Sie die in der Abb. 1 gezeigten Moleküle:
Abb. 1: 1,1'-Diethyl-4,4'-cyanin-Iodid (oben), 1,1'-Diethyl-4,4'-carbocyanin-Iodid (Mitte), 1,1'-Diethyl-4,4'-dicarbocyanin-Iodid; im weiteren Text als Cyanin, Carbocyanin und Dicarbocyanin bezeichnet. Das Iodid-Ion ist nicht eingezeichnet. Der markierte Bereich entspricht einem Polyen aus konjugierten Doppelbindungen. Diese Verbindungen werden auch als Polymethine bezeichnet. Sie bilden eine wichtige Gruppe organischer Farbstoffe. In der Abbildung ist jeweils nur eine der beiden Grenzstrukturen gezeigt.[Clayborn2017]
Zu dem für die Absorption von Strahlung im sichtbaren Bereich verantwortlichen \(\pi\)-Elektronensystem tragen neben den Kohlenstoff-Atomen des Stranges auch die endständigen Stickstoff-Atome bei (sie gehören dazu). Es handelt sich um die farbig markierten Bereiche in der Abb. 1. Da eines der beiden Stickstoffatome zwei Elektronen beiträgt, ist die Gesamtzahl \(Z_{\pi}\) der in diesem Teilbereich des Moleküls enthaltenen π-Elektronen gleich der Anzahl der darin enthaltenen Kohlenstoff- und Stickstoffatome + 1, also gleich der Gesamtzahl \(N\) der Atome längs der Kette +1: \(Z_{\pi}=N+1\). Im Cyanin (\(N=9\), Abbildung 1 oben) weist die Kette also 10 \(\pi\)-Elektronen auf, im Carbocyanin (Abb. 1 Mitte, N=11) sind es 12 \(\pi\)-Elektronen und im Dicarbocyanin (Abb. 1 unten, N=13) sind es 14 \(\pi\)-Elektronen.
2.– Während im allgemeinen aufwändige Molekülorbital-Modelle verwendet werden müssen, um das Absorptionsverhalten von Farbstoffen zu beschreiben, lässt sich auf die Cyanine mit Erfolg ein äußerst einfaches Modell anwenden: das Modell der Teilchen im eindimensionalen Kasten.
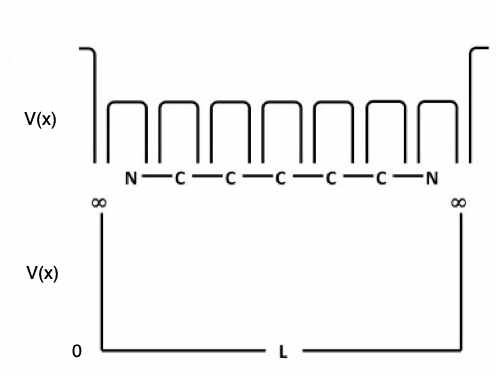
In der Abb. 2 sind zwei Potentialmodelle für Cyanin-Farbstoffe schematisch dargestellt.
Abb. 2: Schematische Modelle des elektrostatischen Potentials, dem die \(\pi\)-Elektronen in Cyanin-Molekülen ausgesetzt sind. Oben: Das Potential variiert längs der Kette (die eingezeichnete Variation ist aber nicht realistisch). Unten: das Potential ist im Bereich der gesamten Kette stets gleich Null und außerhalb der Kette unendlich hoch. Dies ist das Modell des eindimensionalen Kastens. Beachten Sie, dass die Länge \(L\) des Kastens nicht gleich der Summe der atomaren Abstände ist, sondern darüber hinausgeht – dies wird in Abb. 3 erklärt. (vgl. Abb. 3).
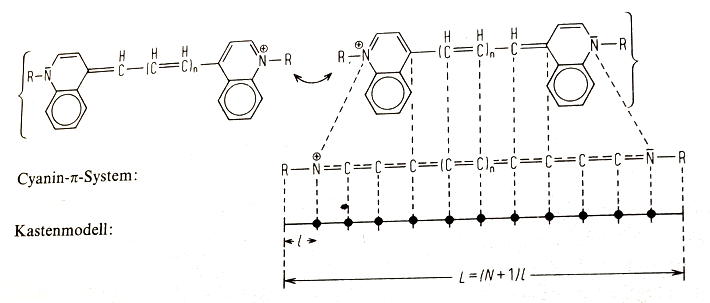
Abb. 3: Während das System konjugierter Doppelbindungen aus insgesamt \(N\) Atomen durch die beiden endständigen Stickstoff-Atome abgeschlossen wird, geht die Länge des eindimensionalen Kasten darüber hinaus: auf diese Weise wird berücksichtigt, dass das \(\pi\)-Elektronensystem über das Zentrum der endständigen Stickstoffatome hinausreicht. Ist \(\ell\) der als konstant angenommene Abstand benachbarter Atome in der Kette, dann ist die Gesamtlänge des Kastens nicht gleich der Summe der Abstände \(\ell\), sondern am Rand der Kette kommt beidseits jeweils noch ein Abstand \(\ell\) hinzu. Die Gesamtlänge \(L\) des Kastens entspricht damit der Zahl \(Z_{\pi}\) der \(\pi\)-Elektronen, multipliziert mit \(\ell\): \(L=Z_{\pi} \cdot \ell = \left(N+1\right)\cdot \ell.\) Näheres siehe Ref. [Heilbronner1978]
Hinweis: beachten Sie, dass die Anzahl der interatomaren Abstände \(\ell\) um eins kleiner ist als die Anzahl der Atome in der Kette. Für Cyanin ist die Zahl der Atome in der Kette gleich 9, die Zahl der interatomaren Abstände aber gleich 8. Am rechten und linken Ende der Kette kommt jeweils ein Abstand \(\ell\) hinzu, also ist die Anzahl der Abstände \(\ell\) gleich 10 und damit gleich der Anzahl der \(\pi\)-Elektronen.
Das Modell führt in vielen, aber nicht in allen Fällen zu einer erfolgreichen Prognose. Der wesentliche Grund für Abweichungen besteht darin, dass der Potentialverlauf im Bereich des \(\pi\)-Elektronensystems nur in grober Näherung approximiert wird. Im Molekül bilden die Kerne ein periodisches Störpotential, das im Modell des eindimensionalen Kastens vernachlässigt wird.
3.– Teilchen im Kasten.– Die ausführliche Durchführung der Berechnung der Energiezustände in einem Kasten entsprechend Abb. 2 (unten) ist in Lehrbüchern der Physikalischen Chemie dokumentiert, beispielsweise in [ref Wedler]. Hier werden nur die wesentlichen Schritte angegeben.
Die zeitunabhängige Schrödinger-Gleichung lautet für eine Dimension (\(x\)-Achse):
\begin {equation} \label {eqSG}
- \frac{\hbar^2}{2m} \frac{{\rm d}^2\Psi}{{\rm d}x^2} + V(x) = E \cdot \Psi.
\end {equation}
Für das Teilchen im eindimensionalen Kasten gelten die Randbedingungen[1]:
\begin{align*}
V&=0\; {\rm für}\;\; 0 < x < L\\
V&=\infty\; {\rm für}\;\; x \leq 0 \; {\rm und}\;\;x \geq L.
\end{align*}
Aus den Randbedingungen folgt für die Lösungsfunktionen von Gl. \ref{eqSG}:
\begin{align*}
\lim_{x\to 0^+}\Psi&=0 ;\\
\lim_{x\to L^-}\Psi&=0;\\
\Psi&=0\; {\rm für}\;\; x \leq 0;\\
\Psi&=0\; {\rm für}\;\; x \geq L;\\
\end{align*}
Eine mögliche relle Lösungsfunktion von Gl. \ref{eqSG} mit den genannten Randbedingungen im Inneren des Kastens lautet:
Der Faktor \(\sqrt{\frac{2}{L}}\) normiert die Wellenfunktionen \(\Psi_n\) so, dass
\[
\int_0^L \frac{2}{L} \cdot \sin^2 \left(\frac{n \cdot \pi \cdot x}{L} \right) \cdot {\rm d}x = 1.
\]
Die Energie der durch die Lösungsfunktionen charakterisierten Zustände ergibt sich wie folgt:
\begin{equation} \label{eqEnergy}
E_n = \frac{h^2}{8 \, m \, L^2} \cdot n^2,
\end{equation}
mit der Masse \(m\) des Teilchens und der Planckschen Konstanten \(h\). Das Teilchen ist hier ein Elektron, also ist \(m=m_e = 9.109 \cdot 10^{-31}\,{\rm kg}\).
Für die Polymethine sind die relevanten Teilchen Elektronen, d.h. \(m=m_e=9.109 \cdot 10^{-31}\;{\rm kg}\).
Die Energie \(E_n\) hängt demnach quadratisch von \(n\) ab.
Mir größer werdender Kastenlänge \(L\) sinkt die Energie der Zustände quadratisch ab.
Die Differenz der Energien zweier Niveaus mit den Quantenzahlen \(n\) und \(n'\) ergibt sich zu
\[
\Delta E_{n,\,n'} = \frac{h^2}{8 \, m_e \, L^2} \cdot \left(n'^2 - n^2 \right)
\]
was für benachbarte Niveaus (\(n' = n+1 \)) übergeht in
\begin{equation} \label{eqDeltaEnergy}
\Delta E_{n,\,n'} = \frac{h^2}{8 \, m_e \, L^2} \cdot \left(2n+1\right).
\end{equation}
Die Zahl \(n\) in Gl. \ref{eqDeltaEnergy} bezieht sich auf das untere der beiden benachbarten Niveaus.
Während also die Energie \(E_n\) der Niveaus mit \(n^2\) ansteigt, steigt \(\Delta E_{n,\,n+1} \) linear mit \(n\).
Die Umrechnung in die Wellenlänge einer Strahlung, deren Photonenenergie diesem \(\Delta E_{n,\,n+1}\) entspricht, ergibt sich zu
\begin{equation} \label{eqLambda}
\lambda = \frac {hc}{\Delta E} = \frac{8\,c \cdot m_e }{h} \cdot \frac{L^2}{2n+1}.
\end{equation}
Umgekehrt kann die Kastenlänge \(L\) aus der Wellenlänge ermittelt werden:
\begin{equation} \label{eqDetL}
L = \sqrt{\frac{ \lambda \cdot h } {8\cdot c\cdot m_e}\cdot \left(2n+1\right)}.
\end{equation}
Es ist sehr wichtig, dass die berechnete Länge \(L\) nicht mit der Summe der Abstände der Atome in der Kette übereinstimmt, da das \(\pi\)-Elektronensystem aus der Kette der Atome beidseitig um einen atomaren Abstand \(\ell\) hinausragt. Für die mit Hilfe von Gl. \ref{eqDetL} berechnete Länge \(L\) gilt (vgl. Abb. 3 und Ref. [Heilbronner1978])
\begin{equation} \label{eqLpiStern}
L=Z_{\pi} \cdot \ell = \left(N+1\right)\cdot \ell,
\end{equation}
wenn \(\ell\) der interatomare Abstand und \(N\) die Gesamtzahl der Atome in der Kette (einschl. der beiden endständigen Stickstoff-Atome) ist. Die Länge \(L'\) der linearisierten Kette der Atome des Polyens vom linken zum rechten Stickstoffatom ergibt sich zu
\begin{equation} \label{eqLAtome}
L' = \left(Z_{\pi}-2\right) \cdot \ell.
\end{equation}
Aus Gl. \ref{eqLpiStern} kann \(\ell\) berechnet werden, da \(Z_{\pi}\) für jeden Farbstoff bekannt ist; aus Gl. \ref{eqLAtome} kann die Länge der Kette des Polyens berechnet werden.
Der nach Gl. \ref{eqLpiStern} berechnete interatomare Abstand \(\ell\) muss ungefähr dem Mittelwert einer C-C-Einfach- und einer C=C-Doppelbindung entsprechen.
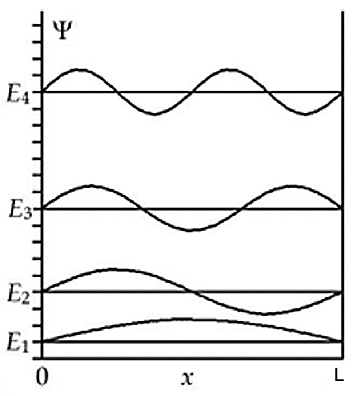
4.– Besetzung von Niveaus mit Elektronen.- Die nachfolgende Abb. 4 zeigt die ersten vier Energieniveaus zusammen mit den zugehörigen Wellenfunktionen.
Abb. 4: Die ersten vier Energieniveaus eines eindimensionalen Kasten mit den zugehörigen Wellenfunktionen. Jedes Niveau kann zwei Elektronen aufnehmen (Pauli-Prinzip). Ein Niveau \(E_0\) mit \(E=0\) existiert nicht, weil die zugehörige Wellenfunktion dann gleich Null wäre und das Teilchen nicht vorhanden wäre. Also hat bereits das tiefste überhaupt existenzfähige Niveau \(E_1\) eine von Null verschiedene Energie. [Kraska2019]
Wenn sich mehrere Elektronen im Kasten befinden, dann müssen diese dem Pauli-Prinzip folgen, d.h. jedes Niveau kann nur durch höchstens zwei Elektronen besetzt werden. Es wird näherungsweise angenommen, dass sich die potentielle Energie im Inneren des Kastens durch die Anwesenheit von Elektronen nicht ändert, es also keine Elektronenkorrelation gibt und sich damit auch die Energieniveaus \(E_n\) gemäß Gl. \ref{eqEnergy} (siehe Abb. 4) durch die Anwesenheit von Elektronen nicht ändert. Der Grundzustand des Systems ergibt sich dann daraus, dass man die Energieniveaus des Kastens von unten nach oben mit jeweils zwei der verfügbaren Elektronen auffüllt. Da die Zahl \(Z_{\pi}\) der \(\pi\)-Elektronen in der Kette gerade ist, sind die Niveaus entweder doppelt besetzt oder leer. Das höchste besetzte Orbital wird als HOMO bezeichnet, das niedrigste unbesetzte als LUMO. Der experimentell beobachtete Übergang findet zwischen dem HOMO und dem LUMO statt (HOMO-LUMO-Übergang).
Die Zahl \(n\) in Gl. \ref{eqDetL} ist gleich dem \(n\) des HOMO. Um das HOMO des jeweiligen Farbstoffes zu finden, muss die Zahl \(Z_{\pi}\) jeweils durch 2 geteilt werden. Für Cyanin ist \(Z_{\pi}=10\) (siehe oben), also ist \(n=5\). Für Carbocyanin ist \(n=\frac{12}{2} = 6\), und für Dicarbocyanin ist \(n=\frac{14}{2}=7\).
5.– (Dieser Punkt entfällt im Rahmen des Farben-Moduls LV 21575) Radiative Übergänge zwischen Zuständen des Teilchens im Kasten sind nur möglich, wenn das Übergangsmoment \(\mu\) von Null verschieden ist (vgl. Lehrbücher der Physikalischen Chemie).
Das Übergangsmoment ist im eindimensionalen Fall für einen Kasten der Länge \(L\) definiert als das Integral
\begin{equation} \label{eqMu}
\mu = e_0 \cdot \int_0^L \Psi_n \cdot x \cdot \Psi_{n'} \;\diff x.
\end{equation}
mit der Ladung \(e_0 = 1.602 \cdot 10^{-19}\;{\rm C}\) des Elektrons.
Für den Übergang \(n=1 \to n=2\) ergibt sich beispielsweise:
\begin{equation} \label{eqMuBeispiel}
\mu = e \cdot \frac{2}{L} \cdot \int_0^L \sin \left(\frac{\pi \cdot x}{L}\right) \cdot x \cdot \sin \left(\frac{2 \pi \cdot x}{L}\right) \;\diff x.
\end{equation}
\(\mu\) hängt also sowohl von \(n\) als auch von der Kastenlänge \(L\) ab.
Eine Rechnung für beliebige \(n\), die hier nicht durchgeführt wird, zeigt, dass Übergänge mit ungeradem \(\Delta n\), also \(\Delta n = \pm 1, \pm3, \pm 5, (\ldots\)), erlaubt sind, während Übergänge mit geradem \(\Delta n\) nicht auftreten können.
Das Betragsquadrat des Übergangsmomentes ist der integralen Extinktion des beobachteten Übergangs proportional.
Das Kastenmodell führt für Carbocyanine zu besseren Ergebnissen als für unsubstitutierte Polymethine; die Ursachen hierfür werden in der Literatur diskutiert [J. Autschbach, J. Chem. Educ. 2007, 84, 11, 1840 (2007)]. In den Cyaninen sind die Atome längs der Kette äquidistant angeordnet; dies führt dazu, dass das Molekül gut durch einen linearen Kasten approximiert werden kann. Dies ist bei den Carbocyaninen der Fall, während die alternierenden Abstände der Atome In unsubstituierten Polymethinen sind die Abstände alternierend kleiner und größer (geringere Delokalisation des \(\pi\)-Elektronensystems), was zu stärkeren Abweichungen vom Modell führt.
Experimentelle Durchführung
Von den Farbstoffen Cyanin, Carbocyanin und Dicarbocyanin (vgl. Abb. 1) stehen Stammlösungen der Konzentration \(c_0=500\;\mu{\rm mol/L}\) im Lösemittel Dimethylsulfoxid (DMSO) bereit. Stellen Sie hieraus jeweils eine 1:100-Verdünnung her, indem Sie \(100\;\mu{\rm L}\) der Stammlösung mittels einer Eppendorf-Pipette in einen Messkolben überführen und dann mit DMSO auf \(10\;{\rm mL}\) auffüllen.
Der zuständige Assistent wird Sie in die Umgangsweise mit dem Spektrometer einweisen.
Es wird für jeden Farbstoff zunächst im Wellenlängenbereich \(300\;{\rm nm} < \lambda < 1000\;{\rm nm} \) die Leertransmission (%T) aufgenommen (Küvette mit DMSO ohne Farbstoff) und abgespeichert.
Nachfolgend wird die Küvette mit der farbstoffhaltigen Lösung gefüllt und erneut die Transmission %T im selben Wellenlängenbereich aufgenommen und abgespeichert. Die binär gespeicherten Daten werden in das ASCII-Datenformat konvertiert und dann auf ein externes Speichermedium überführt. Von dort können sie auf einen Personal Computer (Laptop) übertragen werden.
Die Transmission des Leerspektrums und des Farbstoffes werden durcheinander geteilt und nachfolgend dekadisch logarithmiert, um die Extinktion \(\log \frac{I_0}{I} \) zu erhalten.
Alle Spektren werden in einer gemeinsamen Abbildung dargestellt. Nutzen Sie unterschiedliche Farben und eine geeignete Textbox, um die Spektren der Farbstoff unterscheidbar zu machen. Die Textbox sollte auch die jeweiligen Wellenlängen \(\lambda_{\rm max}\) der Absorptionsmaxima enthalten.
Aufgaben/Auswertung
Die Transmission (%T) des Leerspektrums (\(I_0\)) und des Farbstoffes (\(I\)) werden durcheinander geteilt und nachfolgend dekadisch logarithmiert, um die Extinktion \(E = \log \frac{I_0}{I}\) als Funktion der Wellenlänge zu erhalten.
Die Extinktionsspektren aller drei Farbstoffe werden in einer gemeinsamen Abbildung dargestellt. Nutzen Sie unterschiedliche Farben und eine geeignete Textbox innerhalb der Abbildung, um die Spektren der Farbstoff unterscheidbar zu machen. Die Textbox sollte auch die jeweiligen Wellenlängen \(\lambda_{\rm max}\) der Absorptionsmaxima enthalten.
Berechnen Sie für alle Farbstoffe (Cyanin, Carbocyanin und Dicarbocyanin) aus der Wellenlänge des Absorptionsmaximums \(\lambda_{\rm max}\) die Kastenlänge \(L\) gemäß Gl. \ref{eqDetL}.
Berechnen Sie für alle Farbstoffe (Cyanin, Carbocyanin und Dicarbocyanin) aus der zuvor berechneten Kastenlänge \(L\) den Bindungsabstand \(\ell\) in der Polymethin-Kette gemäß Gl. \ref{eqLpiStern}.
Berechnen Sie gemäß Gl. \ref{eqLAtome} die Gesamtlänge \(L'\) der gestreckt gedachten Polymethinkette.
(Dieser Punkt entfällt, wenn der Versuch im Rahmen des Farben-Moduls LV 21575 durchgeführt wird) Wie kritisch antwortet die Wellenlänge \(\lambda_{\rm max}\) auf eine Änderung der Kastenlänge? Erstellen Sie eine Graphik, in der Sie die nach Gl. \ref{eqLambda} zu erwartende Wellenlänge \(\lambda_{\rm max}\) für Dicarbocyanin als Funktion der Kastenlänge im Bereich \(\frac{L}{2} \leq L \leq 2L\) graphisch darstellen, wobei Sie für \(L\) den von Ihnen errechneten Wert nutzen. Tragen Sie Ihren gemessenen Wert in die Graphik ein und ermitteln Sie daraus die sich rechnerisch ergebende Kastenlänge. Um wieviel nm ändert sich \(\lambda_{\rm max}\), wenn \(L\) um 20% anwächst bzw. abnimmt?
Vergleichen Sie Ihr Ergebnis des C-C-Abstandes \(\ell\) mit dem Mittelwert des Abstandes einer C-C-Einfachbindung und einer C=C-Doppelbindung nach Literaturangaben (Lehrbuch der Organischen Chemie zitieren).
Zu welchem Absorptionsmaximum \(\lambda_{\rm max}\) würde der Mittelwert des Abstandes einer C-C-Einfachbindung und einer C=C-Doppelbindung nach Literaturangaben für die drei Cyanine führen? Wie groß ist die Abweichung vom gemessenen Wert jeweils in nm und in Prozent?
(Dieser Punkt entfällt, wenn der Versuch im Rahmen des Farben-Moduls LV 21575 durchgeführt wird) Berechnen Sie das Verhältnis der Übergangswahrscheinlichkeiten von Cyanin und Dicarbocyanin und vergleichen sie es mit dem Verhältnis der integralen Intensitäten der beobachteten Übergänge der Moleküle.
Das Integral \ref{eqMu} lässt sich am besten numerisch berechnen. Hierzu können Sie die Software Igor Pro oder eine andere geeignete Datenanalyse-Software benutzen.
Zur Berechnung des Integrals mittels Igor Pro erzeugen Sie an der Kommandozeile eine wave (zum Beispiel mit dem Namen w1) und schreiben die Funktion \(\Psi \cdot x \cdot \Psi'\) aus Gl. \ref{eqMu} mit den Werten von \(L\) und n(HOMO) des jeweiligen Farbstoffes hinein:
variable LL=2e-9 // die Kastenlänge (willk. Beispiel)
variable e_0=1.60217733e-19 // Elementarladung in C
variable n_0=5 //Quantenzahl HOMO, Beispiel
make/N=2048 w1
setscale x,0,LL,w1
w1 = e_0 * 2/LL * sin(n_0*pi*x/LL)* x * sin((n_0+1)*pi*x/LL)
display w1
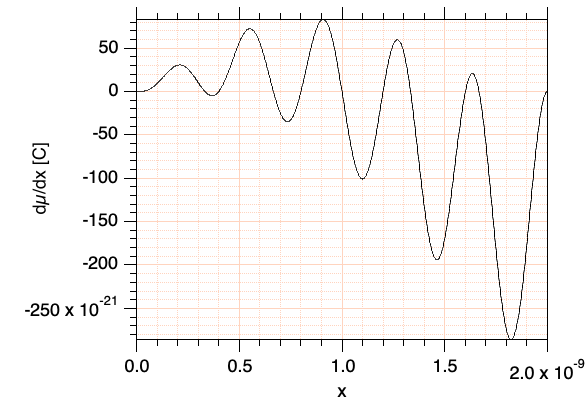
Stellen Sie die in w1 gespeicherte Funktion zur Sicherheit graphisch dar; für die Werte \(L=2 \cdot 10^{-9}\;{\rm m}\) und \(n_0=5\) (Quantenzahl HOMO) sollte sich ungefähr die folgende Abbildung ergeben:
Abb. 4: Graphische Darstellung der Funktion \(\Psi \cdot x \cdot \Psi'\) aus Gl. \ref{eqMu} für \(L=2\;{\rm nm}, n({\rm HOMO})=5\). Die Funktion ist die Ableitung von \(\mu\) nach der Ortskoordinate und hat demzufolge die Einheit Coulomb. Das Übergangsmoment ist gleich der vorzeichenbehafteten Fläche unter der Kurve.
Jetzt können Sie auf ganz einfache Weise das Integral der Funktion, also das Übergangsmoment \(\mu\) von \(x=0\) bis \(x=L\) und sein Quadrat bestimmen:
variable mu=area(w1,0,L)
variable muSquared=mu*mu
print mu
print muSquared
Durch Anpassung von \(L\) und \(n_0\) an das jeweilige Farbstoffmolekül können Sie die entsprechenden Größen erhalten.
Die integrale Absorption der Farbstoffmoleküle können Sie unter Nutzung der Spektren-Graphik ebenfalls mit dem Befehl area ermitteln.
Vergleichen Sie für Cyanin/Dicarbocyanin nun die Verhältnisse der Übergangswahrscheinlichkeiten \(\mu^2\) mit dem Verhältnis der experimentell beobachteten integralen Intensitäten!
Fehlerrechung.–
Es ist der Fehler in \(L\) zu ermitteln. Gl. \ref{eqDetL} kann für die Zwecke der Fehlerrechnung formuliert werden als:
\begin{equation} \label{eqErrAnal}
L = K \cdot \sqrt{\lambda}.
\end{equation}
mit \(K =
\sqrt{\frac{ h } {8\cdot c\cdot m_{e}}\cdot \left(2n+1\right)}\). Die Konstante \(K\) kann als nicht fehlerbehaftet betrachtet werden, denn \(n\) ist eine ganze Zahl, \(h\) und \(c\) sind durch Definition festgelegt und \(m_e\) ist auf 11 Stellen genau bekannt.
Demnach hängt der Fehler \(\Delta L\) nur vom Fehler \(\Delta \lambda_{\rm max}\) ab, den wir dem Auflösungsvermögen des Spektrometers von \(\Delta \lambda = 5\;{\rm nm}\) gleichsetzen.
Demnach ist
\[
\Delta L = \frac{\diff L}{\diff \lambda} \cdot \Delta \lambda = \frac{K}{2\sqrt{\lambda}} \cdot \Delta \lambda.
\]
Literatur: [Clayborn22017]: A. Clayborne, V. Morris, Particle in a Box, Howard University 2017 (Link).
[Heilbronner1978] E. Heilbronner, H. Bock, Das HMO-Modell und seine Anwendung, Verlag Chemie, Weinheim 1978.
[Wedler1982] G. Wedler, H.-J. Freund, Lehr- und Arbeitsbuch Physikalische Chemie, VCH, Weinheim 2018.
[Kraska2019] "Quantenmechanik im Chemieunterricht: Das lineare und das zyklische Kastenmodell angewandt auf Farbstoffe und Aromaten", CHEMKON 2019, 26, Nr. 6, 250 – 260. (Sehr empfehlenswert für Lehramtsstudenten!) LinkHinweise zur Arbeitssicherheit: Sicherheits-Datenblatt Cyanin Sicherheits-Datenblatt Dimethylsulfoxid (DMSO)
Anmerkungen
[1] Die Formulierung der zweiten Randbedingung findet man in vielen Lehrbüchern, aber sie ist streng genommen verkehrt, denn ersten kann ein elektrostatisches Potential als reelle Größe nicht den Wert \(\infty\) annehmen, zweitens ist am Ort \(x=0\) eine unphysikalische Unstetigkeitsstelle, und drittens ist \(\infty\) keine reelle Zahl, die Gleichung funktioniert daher auch mathematisch nicht. Wir beugen uns hier den Gepflogenheiten der Lehrbuchliteratur.
Anleitung zum Speichern von Daten aus dem Spektrometer Shimadzu-UV1280 auf einem externen USB-Speicher: HIER.
Versionen: 14.09.2020 (Erstfassung); 21.05.2023 (Flesch, kleine Änderungen, Link auf Anleitung zum externen Speichern); 24.06.2024: Flesch, kleine Änderungen; Sicherheitsdatenblätter verlinkt.