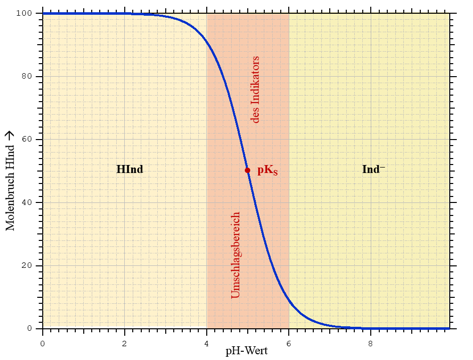
Eine eingehende Diskussion von Indikatorfarbstoffen und ihrem quantitativen Verhalten ist in Referenz [2] zu finden.
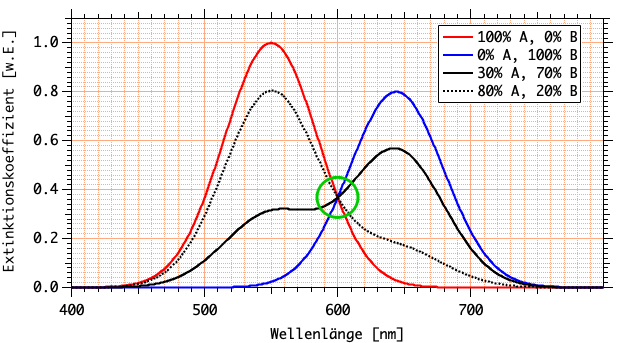
Demnach ist am isosbestischen Punkt nicht nur der Extinktionskoeffizient \(\epsilon\), sondern auch die Extinktion \(E\) beider Substanzen gleich, wenn sie in Abwesenheit der anderen in derselben Konzentration \(c^0\) vorliegen: \[\left(E_{\ce{HInd}} = E_{\ce{Ind-}}\right)_{\lambda = \lambda_{\rm iso}} .\]
Liegt \(\ce{HInd}\) ursprünglich in der Konzentration \(c^0\) vor, und geht \(\ce{Ind-}\) aus \(\ce{HInd}\) hervor, und sind die aktuellen Konzentrationen gleich \([\ce{HInd}]\) und \([\ce{Ind-}]\), dann gilt für die Gesamtextinktion \(E_{\Sigma}\) bei der Wellenlänge \(\lambda=\lambda_{\rm iso}\): \begin{align*} E_{\Sigma} &= E_A + E_B \\ &= \left[\epsilon_{\rm iso} \cdot [\ce{HInd}] \cdot d\right] + \left[\epsilon_{\rm iso} \cdot [\ce{Ind-}] \cdot d \right]\\ &= \left[\epsilon_{\rm iso} \cdot [\ce{HInd}] \cdot d\right] + \left[\epsilon_{\rm iso} \cdot (c^0 - [\ce{HInd}]) \cdot d \right]\\ &= \epsilon_{\rm iso} \cdot [\ce{HInd}] \cdot d + \epsilon_{\rm iso} \cdot c^0 \cdot d - \epsilon_{\rm iso} \cdot [\ce{HInd}] \cdot d\\ &= \epsilon_{\rm iso} \cdot c^0 \cdot d. \end{align*}
Eine beliebige Mischung aus \(\ce{HInd}\) und \(\ce{Ind-}\) wird am isosbestischen Punkt \(\lambda = \lambda_{\rm iso}\) dieselbe Extinktion \(E = E_{\rm iso}\) aufweisen wie \(\ce{HInd}\) und \(\ce{Ind-}\) mit der Konzentration \(c^0\) allein, solange nur insgesamt \([\ce{HInd}] + [\ce{Ind-}] = c^0\).
- Subtraktion einer Konstante vom experimentell ermittelten Spektrum des Gemischs, so dass die langwellige Asymptote eine Extinktion von Null aufweist (die Substanz absorbiert dort nicht).
- Nachfolgende Multiplikation mit einem Faktor, so dass sich das Spektrum im isosbestischen Punkt mit den Spektren der Grenzfälle A und B schneidet.
Erst die Normierung auf den isosbestischen Punkt erlaubt den Vergleich der Spektren!
Gl. \ref{eqk1k2} ist zusammen mit Gl. \ref{eqpKs} die Grundlage zur Auswertung der Messdaten und zur Bestimmung des \(pK_S\)-Wertes des untersuchten Indikatorsystems.
Durchführung
- Wägen Sie ungefähr 25 mg des Indikatorfarbstoffes Thymolblau ab, notieren Sie die genaue abgewogene Menge, überführen Sie die abgewogene Menge in einen 50-mL-Messkolben und füllen Sie mit Ethanol bis zum Eichstrich auf.
- Überführen Sie mit einer Pipette[1] 0,5 mL der hergestellten Lösung in ein Becherglas und fügen Sie mit einer Pipette 19,5 mL Pufferlösung pH=9 hinzu. Füllen Sie eine Quarz-Küvette[2] mit der Lösung und stellen Sie diese in den Probenraum des Spektrometers (vordere Halterung, »Probe«).
- Stellen Sie eine mit Pufferlösung gefüllte Küvette ohne Farbstoff in den hinteren Schacht im Probenraum (»Referenz«).
- Nehmen Sie das Absorptionsspektrum im Modus %T[2] im gesamten zugänglichen Wellenlängenbereich (200 nm < λ < 900 nm) auf und speichern Sie als Text-Datei ab. Dieses Spektrum nennen wir das Probespektrum.
- Füllen Sie die Probenküvette mit der Ethanol/Pufferlösung ohne Indikatorfarbstoff und in demselben Konzentrationsverhältnis Ethanol/Puffer und verankern Sie die Küvette im Probe-Strahlengang. Die Referenzküvette bleibt im Referenzstrahl. Nehmen Sie das %T-Spektrum auf und speichern Sie es als Textdatei ab. Dieses Spektrum nennen wir das Referenzspektrum.
- Lesen Sie beide Spektren in eine Datananalyse-Software (Beispiel: IGOR PRO) ein. Teilen Sie das Referenzspektrum durch das Probespektrum und logarithmieren Sie das Resultat dekadisch. Sie erhalten auf diese Weise die Extinktion der Probe im Bereich 200 nm < λ < 900 nm.
- Nehmen Sie Spektren bei den pH-Werten 0, 1, 2, 3, 6, 8, 9 und 12 auf (jeweils wie oben gezeigt herstellen!). Stellen Sie jeweils die Extinktion \(E\) als Funktion der Wellenlänge graphisch dar.
- Stellen Sie für den pH-Bereich 6-12 die Spektren in derselben Abbildung graphisch dar, bestimmen Sie aus den Spektren mit pH=6 und pH=12 die Wellenlänge des isosbestischen Punktes und normierten Sie die Spektren mit pH=8 und 9 in der oben angegebenen Weise auf den isosbestischen Punkt.
- Wiederholen Sie diese Vorgehensweise fur den pH-Bereich 0–6.
Auswertung
Die Auswertung Ihrer Spektren erfolgt über ein Datenverarbeitungsprogramm, z. B. IGOR PRO.Eine Kurzfassung der Datenanalyse unter Nutzung von Igor Pro wird im Folgenden gegeben:
- Lesen Sie die Spektren in das Programm ein.
- Normieren Sie die Spektren auf den gemeinsamen isosbestischen Punkt.
- Ermitteln Sie entsprechend Gl. \ref{eqk1k2} die relativen Beiträge der Grenzfälle zum Spektrum eines Gemisches.
- Bestimmen Sie hieraus gemäß Gl. \ref{eqpKs} den \(pK_S\)-Wert des Indikatorsystems.
- Vergleichen Sie Ihr Ergebnis mit Literaturdaten. (2)
https://pcprakt.userpage.fu-berlin.de/SKRIPT/T12/AuswertungT12.html
Literatur
(1) H. R. Christen, Grundlagen der Allgemeinen und Anorganischen Chemie, Salle-Sauerländer, Frankfurt/Main 1986, sowie viele andere Lehrbücher der Allgemeinen Chemie.(2) G. Jander und K.-F. Jahr, Maßanalyse, 18. Auflage, de Gruyter, Berlin 2021.
Anmerkungen
[1] Die Pipette wird senkrecht an die schräg gehaltene Wandung des beabsichtigten Aufnahmegefäß gehalten und die Flüssigkeit abgelassen. Hierbei beträgt die Wartezeit nach Ablassen der Flüssigkeit ca. 15 Sekunden. Am Ende wird die Pipette wieder an der Glaswand abgestreift. Da die Pipette auf Auslauf geeicht ist, ist der in der Spitze verbliebene Flüssigkeitsrest nach Ablauf bei der Eichung berücksichtigt worden. NICHT ausblasen!! ↩
[2] Die Küvetten weisen zwei mattierte und zwei transparente Flächen auf. Die transparenten Flächen müssen in den Strahlengang gestellt werden.↩
[3] Unter %T ist das Verhältnis \(\frac{I}{I_0}\) zu verstehen. Diese Größe ist nicht logarithmiert!↩
Version: 03.11.2021 (Freudenberg, Flesch)